All about Dementia

Dementia does not specify cause and instead accompanies many neurological disorders such as neurodegenerative disorders (e.g., Alzheimer’s, fronto-temporal dementia, dementia with Lewy bodies), cerebrovascular disease (e.g., strokes, aneurysms), traumatic brain injuries, multiple sclerosis, infectious and inflammatory disorders, metabolic disorders, hydrocephalus, and brain tumors.
The most common forms of dementia are reviewed here and include Alzheimer’s disease, vascular dementia, frontotemporal dementia, dementia with Lewy Bodies, Parkinson’s disease dementia and Wernicke-Korsakoff Syndrome (WKS). It is important to note that any of these forms of dementia may occur together with one or more of the other forms of dementia.
Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is the most common form of dementia (60-80% of cases) and is considered a “cortical” dementia. That is, individuals with AD have problems in the outer “crust” of the brain called the cerebral cortex. Alzheimer’s disease usually has a slow onset and slow progression of symptoms.
Signs and Symptoms of Alzheimer’s Disease
Patients and their caregivers may notice problems with episodic recent memory manifested in repeating questions and forgetfulness about recent events. Tasks that were relatively easy may slowly become more difficult, particularly more complex cognitive tasks.
As the disease progresses, there is increasing deterioration of memory, language, visuospatial function, and executive abilities. An inability to name familiar objects or people (anomia), misplacing items, getting lost in familiar places, disrupted sleep, incontinence, difficulty swallowing, and depressed mood, agitation, apathy, and psychotic symptoms (delusions, hallucinations) may also occur as the disease progresses. Dependency on caregivers increases as cognition declines.
Tasks or activities of daily living (ADLs) eventually become difficult, including the ability to prepare food, to choose appropriate clothing, and, particularly, to drive. Additionally, the individual’s ability to recognize danger and to accurately and appropriately judge a situation is diminished. Reading and writing become more difficult, and strategies such as making lists may become less effective.
Verbal communication also suffers as the disease progresses, and language becomes confused, with incorrect word usage and mispronunciation of words. A sense of “self” is commonly lost in those with advancing AD.
For additional information, visit the Alzheimer’s Association website at www.alz.org.
Pathology of Alzheimer’s Disease
Amyloid plaques and neurofibrillary tangles are the classic pathological findings in the brain of patients with AD. These plaques and tangles cause degeneration of cells throughout the cerebral cortex, especially the frontotemporal regions.
Amyloid plaques are abnormal accumulations of amyloid proteins between neurons, which disturb the ability of neurons to communicate with each other effectively. Neurofibrillary tangles (NFTs) are fibrous inclusions within the cytoplasm of neurons. The primary component of these NFTs are tau proteins. Tau proteins are associated with microtubules, which are long filaments that help maintain cellular structure. In NFTs, these tau proteins are abnormally phosphorylated and aggregate with other proteins such as ubiquitin.
In early stages of the disease, NFTs are found in the entorhinal cortex, which is an area of the brain associated with olfaction (i.e., sense of smell). As the disease progresses, other areas become affected such as the hippocampus, an area of the brain associated with consolidation of memories. Neurochemically, acetylcholine-producing neurons in an area of basal forebrain, called the nucleus basalis of meynert, are particularly susceptible to damage by amyloid plaques and NFTs (see figure below).

Risk Factors for Alzheimer’s Disease (AD)
- Advancing age
- Family history of AD
- Diabetes
- Obesity
- Untreated hypertension
- High cholesterol
- Stress
- Sedentary lifestyle
- History of head trauma
- History of hypoxic brain injury
- Depression
- Bipolar disorder
- Post-traumatic stress disorder (PTSD)
Genetic Risk Factors
Genetic contributors to AD consist of risk genes and deterministic genes. Deterministic genes are those that can directly cause disease. Three deterministic genes are known to directly cause autosomal dominant Alzheimer’s disease (ADAD):
APP, found on chromosome 21
Presenilin-1 (PS-1) on chromosome 14
Presenilin-2 (PS-2) on chromosome 1
In ADAD, symptom onset is likely to occur before age 60 (it can occur as early as the 30s). Although ADAD is of concern, only about 5% of AD cases are familial. The risk gene with the greatest influence on disease development is the gene for apolipoprotein E (ApoE). ApoE is normally a component of very-low-density lipoproteins (VLDLs). These lipoproteins remove excess cholesterol from the blood and carry it to the liver for degradation. The presence of the gene for the E4 form of this (APOEe4) increases risk; inheritance of this form from both parents increases risk further and may lead to earlier onset of the disease.
Down Syndrome and Alzheimer’s Disease
AD is closely linked to trisomy 21 (Down Syndrome). By the age of 30 to 40, most patients with Down syndrome will develop the plaques and tangles that are associated with AD. These changes are nearly universal among patients with Down syndrome who reach this age, and although the severity of plaque and tangle accumulation mimics that found in AD, not all such individuals will develop AD. One of the possibilities for the connection is that patients have three copies of the APP gene, which is located on chromosome 21.
Management of Alzheimer’s Disease (AD)
Behavioral (non-medication) interventions are essential, effective, and first-line recommendations in the management of AD.
BEHAVIORAL INTERVENTIONS
→ Correcting any sensory deficits (e.g., hearing aids, eye glasses)
→ Making sure the home is safe by rearranging anything thatmight cause falls (e.g., rugs, slippery mats, etc.)
→ Providing well balanced meals with appropriate caloric intake and hydration with fluids and electrolytes
→ At least 25-30min per day of very mild physical activity with assistance (walking around the house, going outside for a short walk)
→ Minimizing daytime naps
→ Keeping lights on and window shades open during the day and lights off and window shades closed at night. This will promote appropriate sleep-wake cycles.
→ Keeping mentally stimulated during the day by doing puzzles, crosswords, coloring, reading, etc.
→ Improving sleep by utilizing sleep hygiene techniques (No bright screens before bedtime, no TV in bed, minimize caffeine, make a consistent sleep schedule and/or routine)
→ Minimizing the use of alcohol and other substances as these can have negative effects on mood and cognition
MEDICATIONS
While there are no medications to date that have reliably reversed or prevented AD, there are medications that may slow down progression of the disease.

Cholinesterase Inhibitors: Recall that Alzheimer’s disease is associated with loss of cholinergic (acetylcholine) neurons, which are important in memory formation. Acetylcholine neurotransmission can be enhanced in many ways. Pharmacologically, the most common method is to inhibit the enzyme that breaks down acetylcholine, acetylcholinesterase. It seems reasonable to assume that these medications would only prove beneficial in patients who still have adequate cholinergic activity. That is, you can’t promote a neurotransmitter if no neurotransmitter is there to begin with.
NMDA Receptor Antagonists: As previously mentioned, glutamate is the most abundant neurotransmitter in the human brain and high concentrations can be toxic to neurons. When neurons die, they can release intracellular glutamate into the extracellular environment. The glutamate that is released can act on nearby glutamate receptors located on other neurons. When NMDA receptors are over-activated by glutamate, calcium channels within the NMDA receptor open and allow a large influx of calcium into the neuron which then induces apoptosis and neuron cell death via biochemical mechanisms that are beyond this discussion. Therefore, NMDA receptor antagonist medications like the noncompetitive NMDA receptor antagonist Memantine (Namenda) have been developed to “protect” neurons from NMDA receptor over activation and destruction. Unfortunately, Memantine has shown mixed results in clinical trials but remains an important medication often co-prescribed with acetylcholinesterase inhibitors.
Other treatments being studied include metal Ion Chelators, antioxidants, and anti-inflammatory agents
Vascular Dementia
Vascular dementia accounts for 20–30% of dementias and often co-occurs with Alzheimer’s Dementia. Vascular dementia can be caused by strokes, heart attacks or any ischemic event that disrupts normal blood flow to (or within) the brain. Impaired blood flow leads to cellular death and damage to brain tissue due to lack of oxygen and nutrients. Symptoms depend on the areas of the brain most affected.
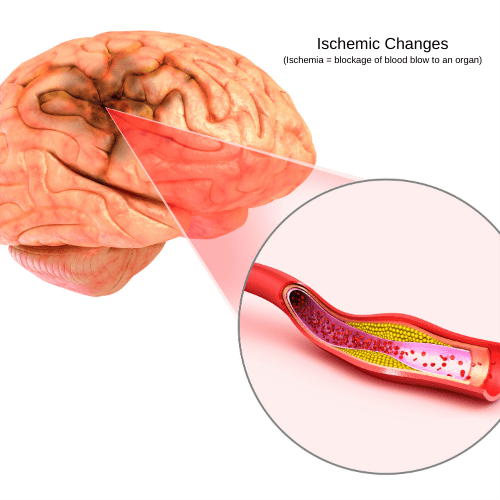
Most common symptoms include
→ Slowed reaction time
→ Poor concentration
→ Poor decision making
→ Confusion
→ Memory impairment
→ Anxiety
→ Hallucinations
→ Ambulation/motor problems
→ Personality changes
Individuals with vascular dementia usually remain functionally stable for a period of time and then suddenly decline in a step-like manner (see graph below).

Fronto-Temporal Dementia (FTD)
Fronto-Temporal Dementia (FTD) accounts for 5–10% of patients with dementia and, as the name implies, is characterized by progressive damage to the frontal and temporal lobes.
Symptoms depend upon the frontal and temporal cortical areas that are affected, but most patients display the following symptoms:
→ Changes in personality: Loss of inhibitory behaviors, socially unacceptable behaviors, aggressiveness
→ Mood changes: Marked lack of empathy with others and mood swings
→ Language problems: Difficulties in finding the correct word to describe something with prominent circumlocution, which is using many words to try and explain something. Also deficits in spontaneous speech.
→ Memory problems: Usually memory deficits occur later in the progression of the condition.
FTD is genetically linked in about 30–50% of cases. The gene for tau protein (discussed above) is most commonly affected. The treatment of FTD is symptomatic and supportive. At present, there is no cure or means of slowing down its progression.
Dementia with Lewy Bodies (DLB) and Parkinson’s Disease Dementia (PDD)
Dementia with Lewy Bodies (DLB) is also referred to as Lewy Body Dementia and accounts for about 3–5% of dementias. LBD and Parkinson’s Disease Dementia (PDD) are very similar, and both are associated with the presence of Lewy Bodies (LBs) in the brain. Lewy Bodies are abnormal proteins that have aggregated or “clumped” together. These protein aggregates contain the protein 𝛼-synuclein and also play a role in Parkinson’s Disease (PD) with and without dementia. However, patients with DLB do not necessarily have PD, although some patients may have both conditions.
The symptoms of DLB are similar to that of Alzheimer Disease but patients tend to develop attention problems, disordered movements, visuospatial deficits, and visual hallucinations earlier in the course of the illness. Parkinson’s Disease Dementia (PDD) is slightly different than Dementia with Lewy Bodies in that the movement disorder (i.e., slowed movements and pill-rolling tremor) usually presents before the cognitive deficits. Currently there is no cure for DLB or PDD, and the treatment is supportive. However, recent clinical trials have suggested that the drugs used in the treatment of Alzheimer’s disease (acetylcholinesterase inhibitors and glutamate receptor antagonists) may be beneficial in alleviating some of the memory and cognitive problems associated with DLB and PDD. For more information about Parkinson Disease, click here.
Wernicke-Korsakoff Syndrome (WKS)
Korsakoff’s Syndrome (KS) is usually due to a deficiency in thiamine (vitamin B1) that occurs most commonly from malnutrition. Malnutrition due to excessive alcohol intake is the most common cause. Alcohol interferes with the conversion of thiamine into thiamine pyrophosphate, which is its active form. The symptoms of WKS include memory loss, denial that there are any difficulties with memory (lack of insight), problems in acquiring new information and skills, personality changes and inventing convincing stories to fill in gaps in memories (also called confabulation). The lack of thiamine (vitamin B1) causes damage to structures in the brain called mammillary bodies. Mammillary bodies are located in the posterior hypothalamus and are connected to the hippocampus in the medial temporal lobe. Damage to these areas compromise the consolidation of short-term memories into long term memories. Treatment includes withdrawal and abstinence from alcohol and administration of high doses of thiamine.
References
- Ferrando, J. L. Levenson, & J. A. Owen (Eds.), Clinical manual of psychopharmacology in the medically ill(pp. 3-38). Arlington, VA, US: American Psychiatric Publishing, Inc.
- Stahl, S. M. (2014). Stahl’s essential psychopharmacology: Prescriber’s guide (5th ed.). New York, NY, US: Cambridge University Press.
- McCarron, Robert M., et al. Lippincotts Primary Care Psychiatry: for Primary Care Clinicians and Trainees, Medical Specialists, Neurologists, Emergency Medical Professionals, Mental Health Providers, and Trainees. Wolters Kluwer Health/Lippincott Williams & Wilkins, 2009.
- Focus Psychiatry Review, Dsm-5: Dsm-5 Revised Edition by Deborah J. Hales (Author, Editor), Mark Hyman Rapaport (Author, Editor)
- American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Washington, DC.
- Arciniegas, Yudofsky, Hales (editors). The American Psychiatric Association Publishing Textbook Of Neuropsychiatry And Clinical Neurosciences.Sixth Edition.
- Bear, Mark F.,, Barry W. Connors, and Michael A. Paradiso. Neuroscience: Exploring the Brain. Fourth edition. Philadelphia: Wolters Kluwer, 2016.
- Blumenfeld, Hal. Neuroanatomy Through Clinical Cases. 2nd ed. Sunderland, Mass.: Sinauer Associates, 2010.
- Cooper, J. R., Bloom, F. E., & Roth, R. H. (2003). The biochemical basis of neuropharmacology (8th ed.). New York, NY, US: Oxford University Press.
- Higgins, E. S., & George, M. S. (2019). The neuroscience of clinical psychiatry: the pathophysiology of behavior and mental illness. Philadelphia: Wolters Kluwer.
- Iversen, L. L., Iversen, S. D., Bloom, F. E., & Roth, R. H. (2009). Introduction to neuropsychopharmacology. Oxford: Oxford University Press.
- Levenson, J. L. (2019). The American Psychiatric Association Publishing textbook of psychosomatic medicine and consultation-liaison psychiatry. Washington, D.C.: American Psychiatric Association Publishing.
- Mendez, M. F., Clark, D. L., Boutros, N. N. (2018). The Brain and Behavior: An Introduction to Behavioral Neuroanatomy. United States: Cambridge University Press.
- Schatzberg, A. F., & DeBattista, C. (2015). Manual of clinical psychopharmacology. Washington, DC: American Psychiatric Publishing.
- Schatzberg, A. F., & Nemeroff, C. B. (2017). The American Psychiatric Association Publishing textbook of psychopharmacology. Arlington, VA: American Psychiatric Association Publishing.
- Neuroscience, Sixth Edition. Dale Purves, George J. Augustine, David Fitzpatrick, William C. Hall, Anthony-Samuel LaMantia, Richard D. Mooney, Michael L. Platt, and Leonard E. White. Oxford University Press. 2018.
- Stahl, S. M. (2013). Stahl’s essential psychopharmacology: Neuroscientific basis and practical applications (4th ed.). New York, NY, US: Cambridge University Press.
- Stern, T. A., Freudenreich, O., Fricchione, G., Rosenbaum, J. F., & Smith, F. A. (2018). Massachusetts General Hospital handbook of general hospital psychiatry. Edinburgh: Elsevier.
- Whalen, K., Finkel, R., & Panavelil, T. A. (2015). Lippincotts illustrated reviews: pharmacology. Philadelphia, PA: Wolters Kluwer.
- Hales et al. The American Psychiatric Association Publishing Textbook of Psychiatry. 6th
- Goldberg & Ernst. Managing Side Effects of Psychotropic Medications. 1st 2012. APP.
- Benjamin J. Sadock, Virginia A. Sadock. Kaplan & Sadock’s Comprehensive Textbook of Psychiatry. Philadelphia :Lippincott Williams & Wilkins, 2000.
- Ebenezer, Ivor. Neuropsychopharmacology and Therapeutics. John Wiley & Sons, Ltd. 2015.
- Stein, Lerer, and Stahl. Essential Evidence-Based Psychopharmacology. Second Edition. 2012.
- Puzantian, T., & Carlat, D. J. (2016). Medication fact book: for psychiatric practice. Newburyport, MA: Carlat Publishing, LLC.
- Meyer, Jerrold, and Quenzer, Linda. Psychopharmacology: Drugs, the Brain, and Behavior. Sinauer Associates. 2018.
